Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)
 Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация) - это наследственное нарушение обмена меди в организме, приводящее к тяжелому поражению внутренних органов (печень, почки) и нервной системы.
Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация) - это наследственное нарушение обмена меди в организме, приводящее к тяжелому поражению внутренних органов (печень, почки) и нервной системы.
Содержание:
Причиной заболевания служит мутация гена ATP7B, расположенного в 13 хромосоме. Именно этот ген контролирует правильный обмен меди в организме. Снижение белка церулоплазмина, отвечающего за выведение меди, приводит к ее накоплению в различных тканях и органах, что нарушает их правильную работу.
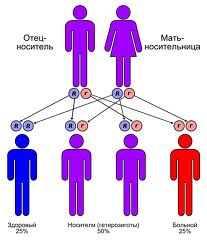
Заболевание наследуется по аутосомно-рецессивному признаку. Болезнь поражает четверть всех братьев и сестер, которые получили по 2 мутантных гена от клинически здоровых родителей (по одному гену от каждого). Дети, которые получили по одному мутантному гену, являются носителями болезни, остаются здоровыми. Чаще болеют представители мужского пола. К счастью, распространенность заболевания не так велика - 3 случая на 100 тыс. населения, если брак близкородственный, то частота увеличивается.
Видео "Болезнь Вильсона-Коновалова"
Заболевание обычно проявляется в 10-26 лет, но возможно появление и в детском, и в более старшем возрасте. Здесь существует такая закономерность: чем раньше возникают симптомы, тем тяжелее протекает само заболевание.
Предположить наличие заболевания у ребенка можно по следующим признакам: боли в области печени, тошнота, горечь во рту; появление на коже груди и спине сосудистых "звездочек"- телеангиоэктазий; желтоватый оттенок склер и кожи; кровоточивость десен; изменение окраски языка -"малиновый, лаковый"; гормональные нарушения (у девушек - нарушения менструального цикла, у юношей - нагрубание грудных сосков, акромегалия); снижение интеллекта и изменение поведения; проблемы с усвоением материала в школе. Течение болезни быстрое - до 3х лет. Так проявляется брюшная форма (чаще поражается печень) болезни Вильсона-Коновалова.
В более старшем возрасте развивается дрожательная форма болезни, течение болезни медленное, до 8-14 лет. В этом варианте отложениями меди поражаются подкорковые узлы головного мозга (чечевицеобразное ядро). Характерны мышечная ригидность и дрожание, сменяющие друг друга. Пациенту становится трудно выполнять мелкие движения, появляется дрожание в конечностях, замедление походки, которое сочетается с повышенным мышечным тонусом. Нарушается речь, она становится замедленной, невнятной, дизартричной; возникают трудности с глотанием. Могут наблюдаться эпилептиформные приступы. С течением времени снижаются память и внимание, появляется депрессия, раздражительность. В тяжелых случаях возникает слабоумие (деменция) и смерть.
 Отложения меди могут обнаруживаться в почках (синдром Фанкони), что нарушает работу почечных канальцев, в сердце (кардиомиопатия), в роговице глаза - кольцо Кайзера-Флейшера (коричнево-зеленый ободок меди по краю роговицы глаза), поражение суставов, поджелудочной железы (сахарный диабет).
Отложения меди могут обнаруживаться в почках (синдром Фанкони), что нарушает работу почечных канальцев, в сердце (кардиомиопатия), в роговице глаза - кольцо Кайзера-Флейшера (коричнево-зеленый ободок меди по краю роговицы глаза), поражение суставов, поджелудочной железы (сахарный диабет).
Специфическая диагностика болезни Вильсона-Коновалова выявляет увеличение экскреции меди с мочой за сутки, снижение белка церулоплазмина и меди в сыворотке крови. Диагностическим признаком служит обнаружение колец Кайзера-Флейшера с помощью щелевой лампы. Важным моментом является сбор семейного анамнеза и генетическое консультирование.
Чем раньше выявлено заболевание и начато лечение, тем лучше эффект. Даже тяжелые неврологические варианты поддаются терапии, вплоть до исчезновения симптомов заболевания. Основу лечения составляет "печеночная"диета (стол 5а) с пониженным содержанием меди. Она исключает шоколад, орехи, кофе, бобовые, субпродукты, ржаной хлеб, грибы, баранина, свинина, некоторые морепродукты.
Для выведения избытка меди используют препараты - комплексоны (тиоловые соединения или соли цинка), которые связывают "свободную" медь и выводят ее из организма. Самыми распространенными являются D-пеницилламин, унитиол, сульфат или оксид цинка.
Дополнительно проводят лечение гепатопротекторами (препараты, улучшающие функцию печени). Если консервативное лечение не помогает, то прибегают к пересадке печени.
Если в семье имеется родственник, страдающий гепатолентикулярной дегенерацией, обязательно проводят ДНК-диагностику заболевания.